Projects
Singlet Fission • Heteroacenes and FETs • Catalysis and Mechanistic Studies • Conjugated Oligomers • Foldamers and Nanographenes • Photodetectors • PAH Dyads
Mechanistic Studies of Organocatalytic Asymmetric Reactions |
Catalysts can be used not only to accelerate chemical reactions but can also to control product distribution. Catalytic processes play an important role towards utilizing our limited resources in an environmentally responsible manner since they can reduce both the energy required for the transformation as well as the amount of chemical waste produced. While catalysts have traditionally been metal-based, the use of organic molecules has recently been gaining interest in part due to its potential to significantly reduce cost and make processes more environmentally friendly, in addition to facilitating unprecedented transformations. However, such organocatalytic processes typically require high catalyst loadings and long reaction times, which can offset their beneficial aspects.
Understanding how a catalyst operates, including how it activates the substrate(s), what is its resting state, whether there are non-productive off-cycle states, and what kinds of non-reversible deactivation pathways are accessible, can provide direction for the development of improved catalysts. Mechanistic studies of asymmetric organocatlytic reactions using state of the art tools and techniques in experimental and theoretical chemistry is shedding light into these processes and providing new catalyst design principles towards developing highly efficient catalytic transformations and being able to activate previously unsuitable substrate classes.
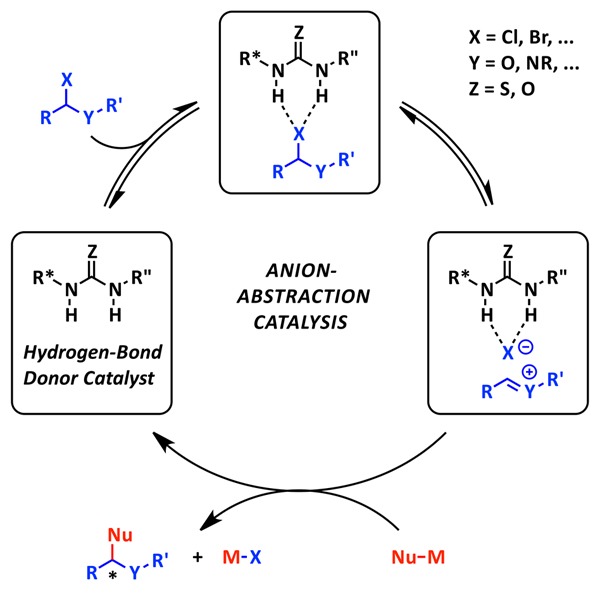
One particularly interesting mode of activation that has recently captured our attention is anion-abstraction catalysis, conceptually shown above. In this mode of catalysis, it has been postulated that the hydrogen-bond donor catalyst abstracts an anion from a neutral organic precursor to generate a cationic electrophile. This reactive ion pair intermediate, now chiral, can then undergo enantioselective attack by a nucleophile to afford the enantioenriched product.
[1] D. D. Ford, D. Lehnherr, C. R. Kennedy, E. N. Jacobsen, J. Am. Chem. Soc. 2016, 138, 7860—7863. [PDF].
[2] D. D. Ford, D. Lehnherr, C. R. Kennedy, E. N. Jacobsen, ACS Catal. 2016, 6, 4616—4620. [PDF].
[3] D. Lehnherr, D. D. Ford, A. Bendelsmith, C. R. Kennedy, E. N. Jacobsen, Org. Lett. 2016,18, 3214—3217. [PDF].
[4] C. R. Kennedy, D. Lehnherr, N. S. Rajapaksa, D. D. Ford, Y. Park, E. N. Jacobsen, J. Am. Chem. Soc. 2016, 138, 13525. [PDF].
Transition Metal Catalysis and Polyheterohalogenated Aromatics |
Transition metal catalysis has enabled countless useful methods to make and break bonds, ultimately reshaping the way chemists think about building molecules. Cross-coupling reactions (e.g., Sonogashira, Suzuki, and Neigishi couplings, Buchwald-Hartwig aminations) are hallmarks of transition metal catalysis and are widely used in industrial settings to make C-C, C-N, C-O, C-S bonds.
Many of these cross-coupling reactions rely on aryl halide precursors, which can be a limiting factor if the necessary aryl halide precursor to a target is not readily available. Despite polyhalogenated aromatics providing highly useful platforms for diversity-oriented synthesis, there are limited methods to access such haloarenes for aromatic cores larger than benzene (e.g. naphthalene). Towards addressing this problem, we have developed a catalytic method to access polyheterohalogenated aromatics, such as polyheterohalogenated naphthalenes with regioselective functionalization of 7 of its 8 positions.[1]
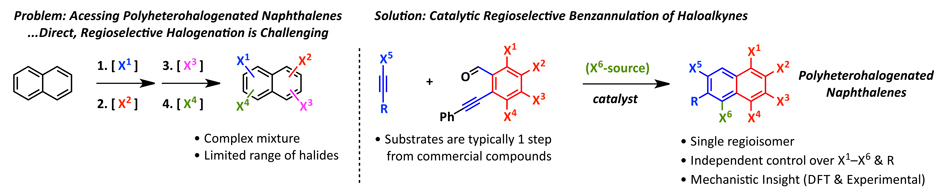
Figure 1. (Left) Illustration of the synthetic problem, polyheterohalogenated aromatics cannot easily be synthesized with good regiochemical control. (Right) Our solution to the problem of how to access polyheterohalogenated aromatics via the regioselective benzannulation of a haloalkyne using a halogenated benzaldehyde in the presence or absence of an additional halide source.
The combination of being able to regioselectivley install different halides and chemoselectivly achieve cross-coupling based on the different reactivity offered by the different halides (e.g. C-Br vs C-Cl bond functionalization) enables polyheterohalogenated aromatics to serve as powerful diverent intermediates. The mechanism was probed experimentally in conjunction with DFT calculations to gain insight into the factors governing reactivity and regioselectivity outcomes. These haloarenes are serving as important precursors to larger aromatic architectures and conjugated polymers, particularly, the incorporation halides into materials is enabling us to tune their properties, including redox potentials, HOMO-LUMO energies, conformational structure, and crystal packing.
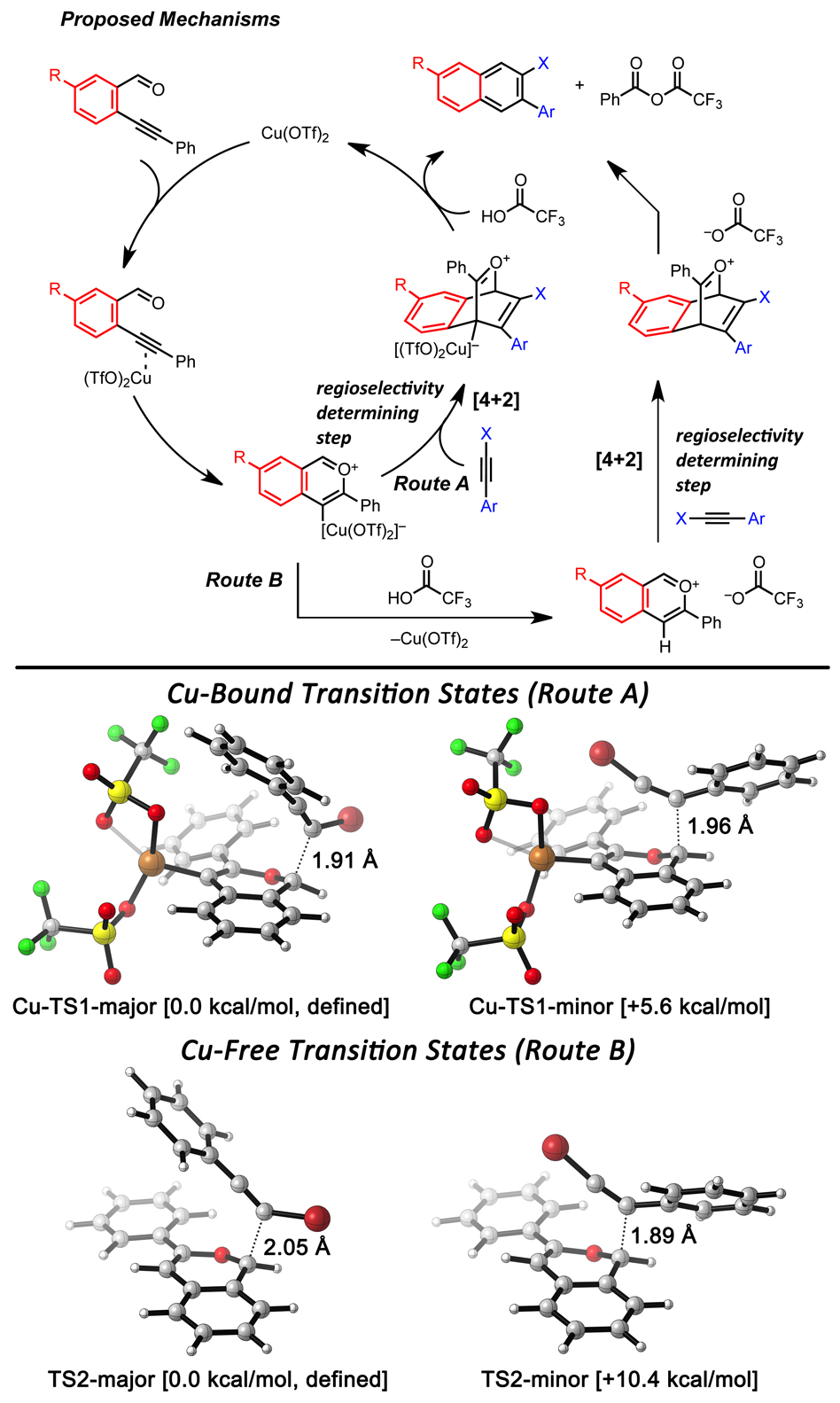
Figure 2. (Top) Proposed reaction mechanisms. (Bottom) DFT calculations of regioselectivity determining transition states via Route A (Cu-bond-TS) or Route B (Cu-Free-TS).
[1] D. Lehnherr, J. M. Alzola, E. B. Lobkovsky, W. R. Dichtel, Chem. Eur. J. 2015, 21, 18122—18127. [PDF].
DFT for the Design & Understanding of Reactions, Catalysts, and Materials |
The ability to predict a priori molecular properties or reactivity aspects, whether they deal with product outcomes arising from different reactivity types or simply regioisomeric products, can be powerful tools in target and reaction design. With the advance of theoretical chemistry and modern computational methods, such as density functional theory (DFT), coupled with the ever increasing computing speed of modern processors and decreasing cost of memory, the ability to bring both understanding and predictivity aspects to ever increasing complex systems is reshaping how we design molecules (e.g., catalysts, materials, pharmaceuticals) as well as the rections and catalysts used to make them.
Case Study 1: In Silico Design of Improved Catalysts
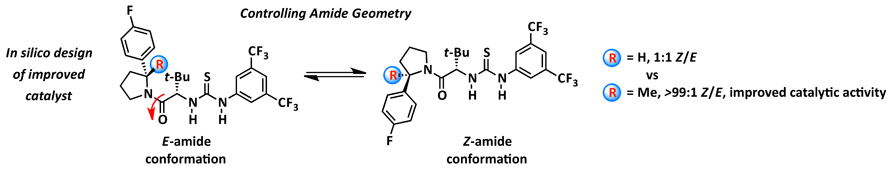
As part of understanding anion-abstraction catalysis in the context of hydrogen-bond donors (HBDs), our mechanistic study revealed that two molecules of catalysts work cooperatively in binding the ion-pair and that catalyst conformation can dramatically affect reaction rate and enantioselectivity. Amide-based HBDs such as the catalyst shown below (R = H) exists in solution as a mixture of slowly interconverting E- and Z-amide rotamers, each with the potential to catalyze reactions with different rates and selectivities. DFT enabled to screen for an appropriate substitution on the pyrrolidine ring to bias the amide rotamer ratio in favor of the more reactive and selective Z-rotamer. Formal methylation of the catalyst (R = H --> Me) provides a catalyst that exists solely as the Z-rotamer and displays improved entioselectivity and reactivity. To further improve catalysis, the enthalpic penalty of having two molecules of catalyst assemble in the transition state around the substrate can be dramatically decreased by covalently linking the two thioureas. The refinement of a DFT model of the transition state structure enabled the in silico design of a covalent linker with appropriate geometric parameters to best match the transition state attainable by the monomeric catalyst (overlay shown below). Ultimately resulting in a dramatically improved catalysis enabling catalyst loading to be lowered to 0.05 mol% while maintaining levels of entioselectivity above 90 %ee.
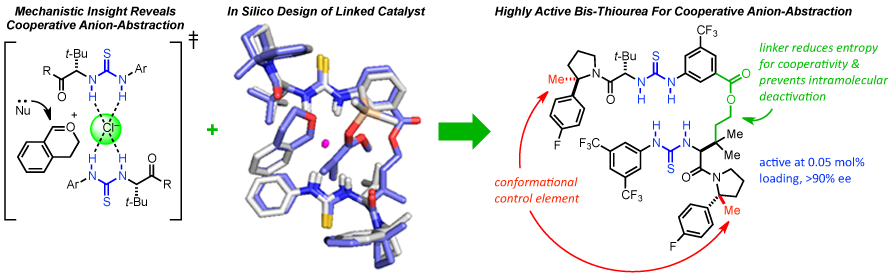
[1] Org. Lett. 2016,18, 3214—3217. [PDF]. [2] J. Am. Chem. Soc. 2016, 138, 13525—13528. [PDF].
Case Study 2: Tailoring Substrates for Site Selectivity & Reaction-Type Selectivity
Cycloaddition reactions can be useful for tailoring the properties of conjugated systems, yet chemo- and regioselective control is not always easily achieved. The use of DFT can be used to predict such reactivity aspects. Towards designing new donor-acceptor materials, DFT was utilized to understand and predict cycloaddition chemistry between TCNE and various pentacene derivatives.
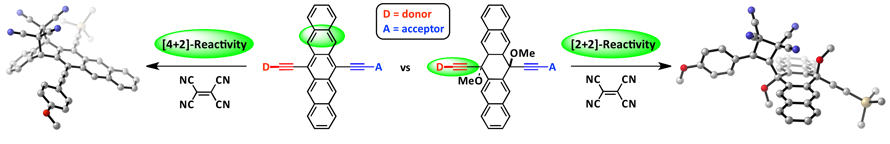
[1] D. Lehnherr, M. Adam, A.H. Murray, R. McDonald, F. Hampel, R. R. Tykwinski, Can. J. Chem. 2017, 95, 303—314. [PDF].
Case Study 3: Elucidation of Reaction Mechanism via Merging DFT and Exerimental Tools
As part developing a Cu-catalyzed method to access polyheterohalogenated naphthalenes from haloalkynes and benzaledehydes, the reaction mechanism was elucidated.
This is described above in the section Transition Metal Catalysis and Polyheterohalogenated Aromatics.